Submit chemicals
In the first step users can upload up to 100 chemicals using SMILES string format or using CASRN ID or DSSTox ID (see EPA comptox). Inputs are standardized, filtered, and mixtures, ions and inorganic chemicals are excluded using the MolVS Python3.6 library.
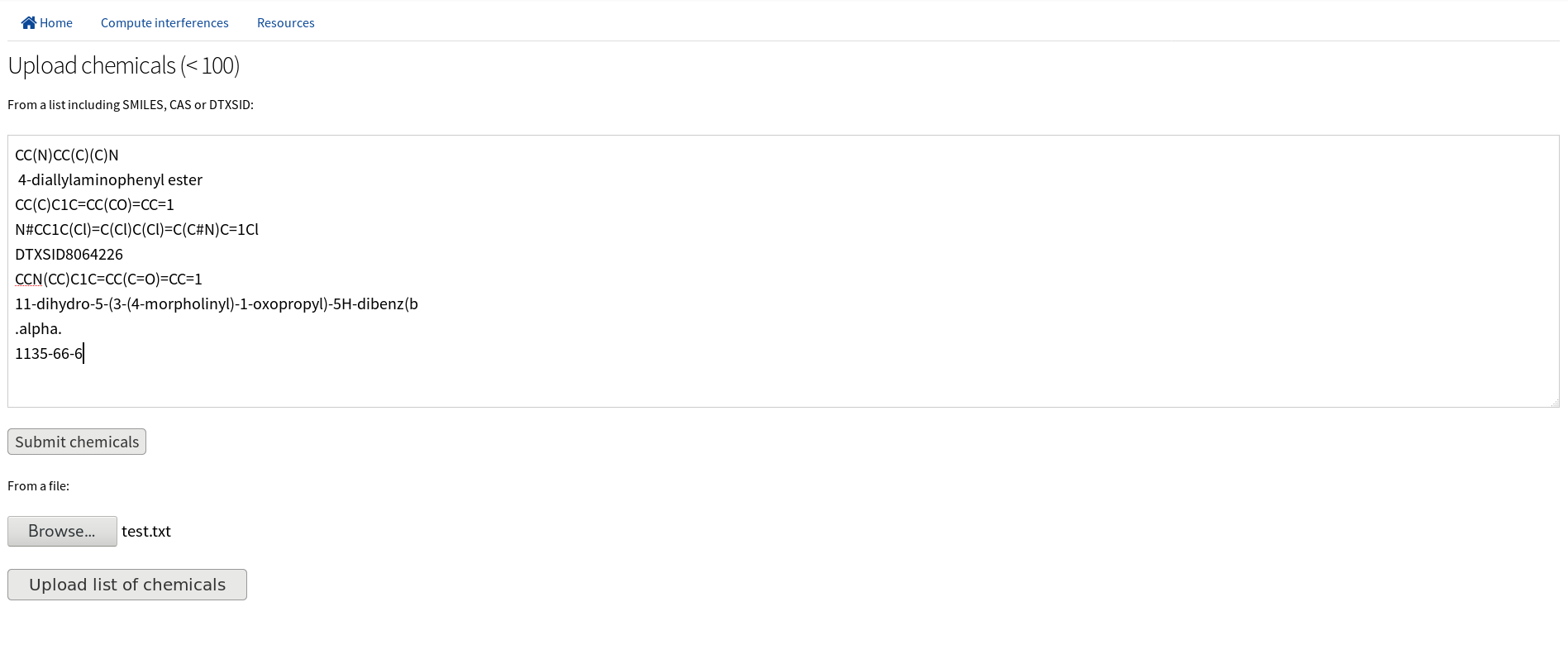
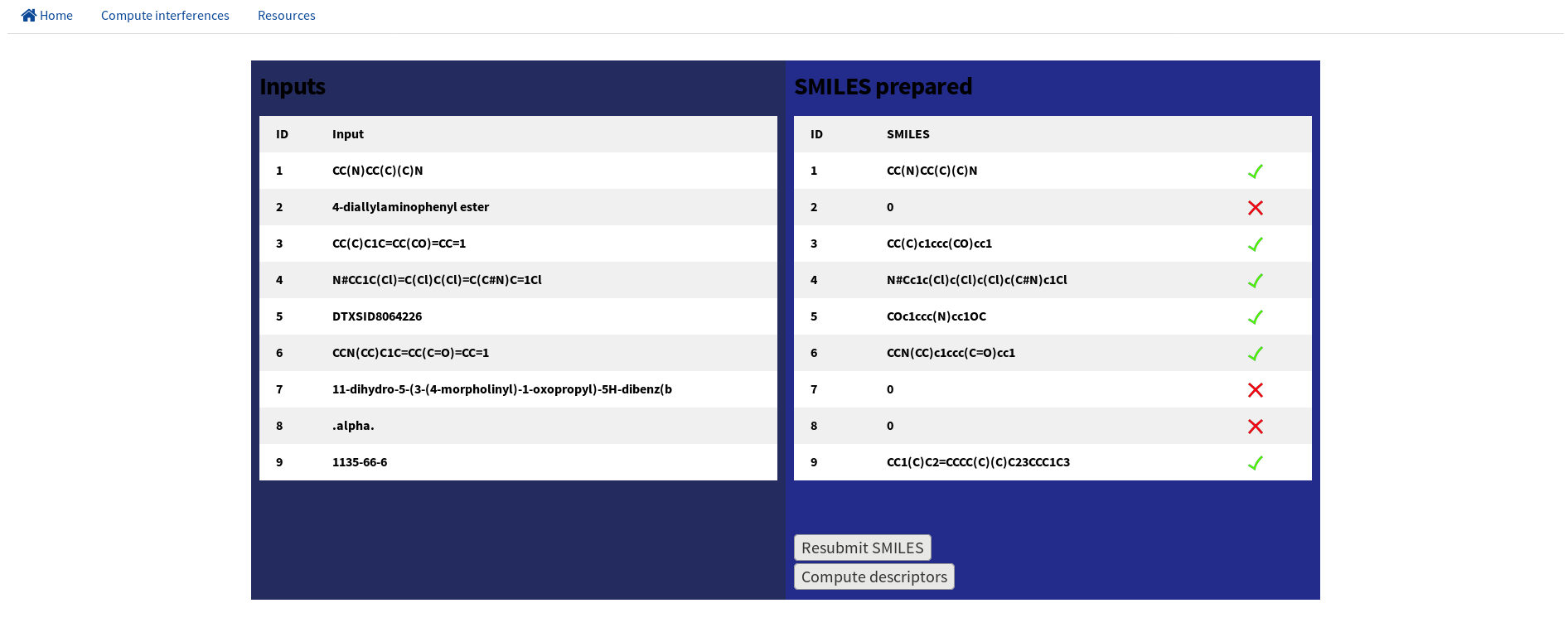
Users can refine the chemical list uploaded at this step.
Compute descriptors
For each chemical a set of 677 structural descriptors is computed including 1D and 2D descriptors as well as physico-chemical descriptors using RDKit tool kit implemented in Python 3.6 and OPERA models. The computational time is around 2-5s per chemical.


Users can download the descriptors matrix in csv format from RDKit tool kit and OPERA model at this step.
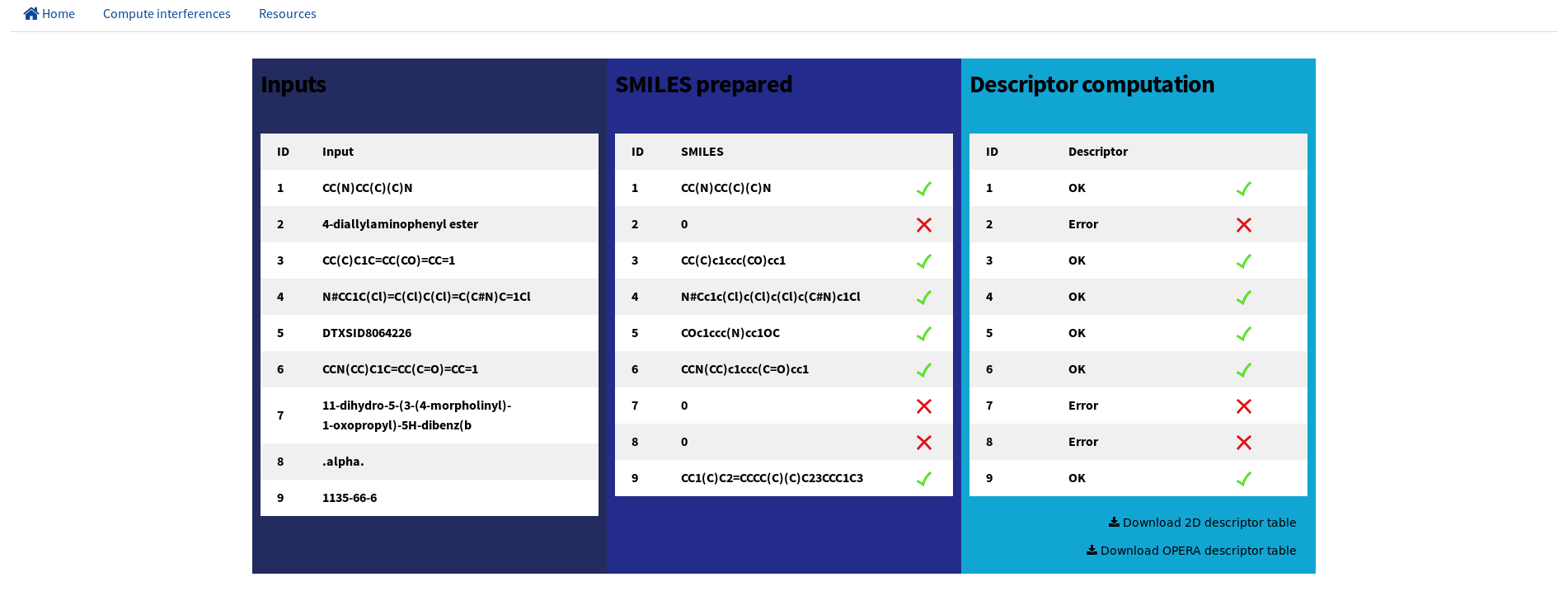
Interference model prediction
Once descriptors have been computed, users can predict chemical interference activity by choosing one or several of the 17 interreference models proposed. Each model is an average of 10 different random forest models built to cover the full set of chemicals (cf. publication). The table below describes the available models.
All random forest models used can be downloaded in a tar.gz archive with the assciated Rscript to run them individually.
It takes around 1-2s per chemical and per model to compute the predictions.

For quality check and speed purpose, each chemical no included in the DSSTox database will be add in our internal database by default. Users can choose to not save chemicals in the databse at this step.
Results
Results are presented in a dynamic table. For each model selected a score between 0 and 1 is reported with an associated standard deviation. The score is a probability for a chemical to interfere with the technology, cell culture and condition related to the model. A score close to 1 signifies that the chemical has a high chance to interfere with that particular technology and experimental condition. The standard deviation is derived from the deviation of the ten random forest model predictions.
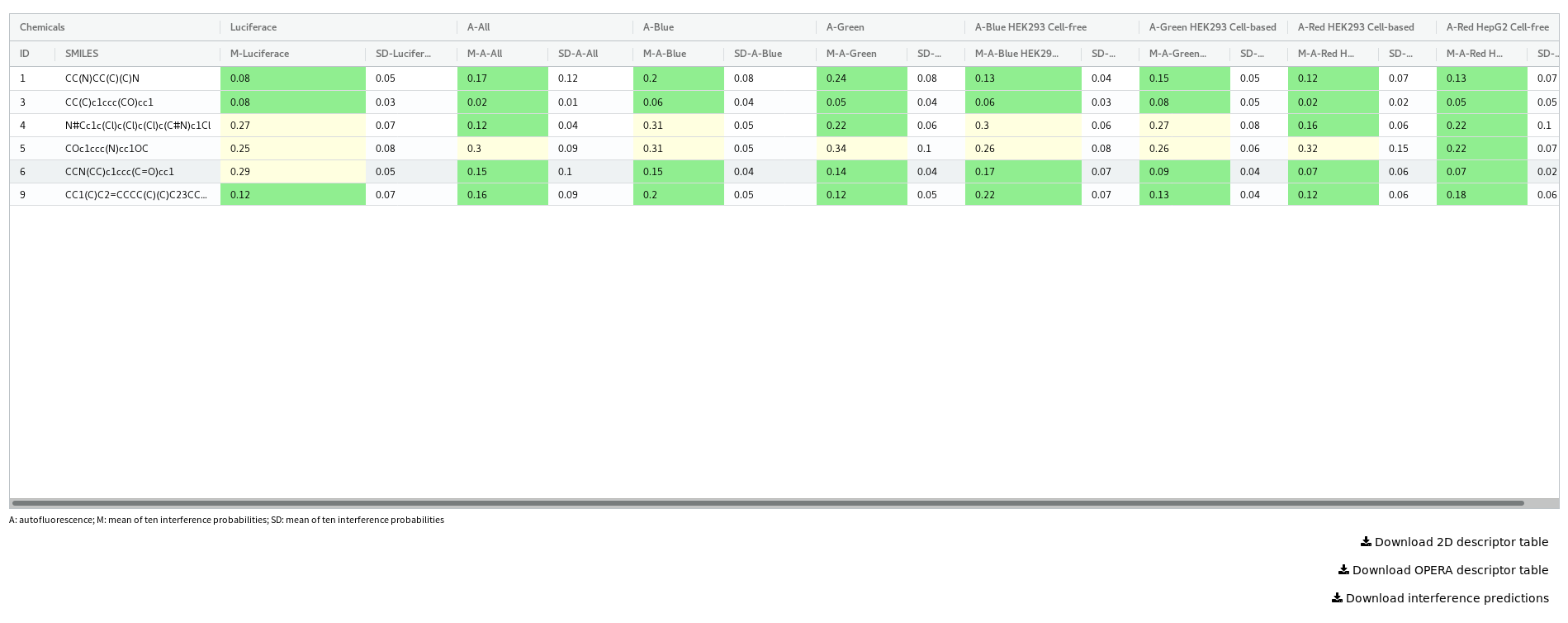
Users can download the table of results in a csv format.
Please cite: A. Borrel et al.; High-Throughput Screening to Predict Chemical-Assay Interference